Table of Contents
- Working with non-animal/plant groups in
phruta - Creating taxonomic constraints in
phruta - Running PartitionFinder in
phruta - Partitioned analyses in RAxML
- Identifying rogue taxa
Working with non-animal nor plant groups in phruta
As explained in the brief intro to phruta, the
sq.curate() function is primarily designed to curate
taxonomic datasets using gbif. Alto gbif is
extremely fast and efficient, it is largely designed to deal with
animals and plants. If you’re interested in using the gbif
backbone for curating sequence regardless of the kingdom use the
following approach:
taxonomy.retrieve(species_names=c("Felis_catus", "PREDICTED:_Vulpes",
"Phoca_largha", "PREDICTED:_Phoca" ,
"PREDICTED:_Manis" , "Felis_silvestris" , "Felis_nigripes"),
database='gbif', kingdom=NULL)Note that the kingdom argument is set to
NULL. However, as indicated in the first vignette,
gbif is efficient for retrieving accurate taxonomy when we
provide details on the kingdom. Given that all the species
we’re interested in are animals, we could just use:
taxonomy.retrieve(species_names=c("Felis_catus", "PREDICTED:_Vulpes",
"Phoca_largha", "PREDICTED:_Phoca" ,
"PREDICTED:_Manis" , "Felis_silvestris" , "Felis_nigripes"),
database='gbif', kingdom='animals')We could also do the same for plants by using plants
instead of animals in the kingdom argument.
Now, what if we were interested in following other databases to retrieve
taxonomic information for the species in our database? The latest
version of phruta allow users to select the desired
database. The databases follow the taxize::classification()
function. Options are: ncbi, itis,
eol, tropicos, nbn,
worms, natserv, bold,
wiki, and pow. Please select only one. Note
that the gbif option in
taxize::classification() is replaced by the internal
gbif in phruta.
Now, let’s assume that we were interested in curating our database
using itis:
taxonomy.retrieve(species_names=c("Felis_catus", "PREDICTED:_Vulpes",
"Phoca_largha", "PREDICTED:_Phoca" ,
"PREDICTED:_Manis" , "Felis_silvestris" , "Felis_nigripes"),
database='itis')Using alternative databases is sometimes desirable. Please make sure you review which the best database is for your target group is before selecting one.
Creating taxonomic constraints in phruta
For different reasons, phylogenetic analyses sometimes require of
tree constraints. phruta can automatically generate trees
in accordance to taxonomy and a backbone topology. We divide constraint
trees into two: (1) ingroup+outgroup and (2) particular clades.
ingroup + outgroup
In this constraint type, phruta will create monophyletic
groups for each of the taxonomic groups in the database (for selected
target columns). Finally, it will generate tree with the same topology
provided in the Topology argument. The user will provide
the species names of the outgroup taxa as a vector of string that should
fully match the names in the taxonomy file.
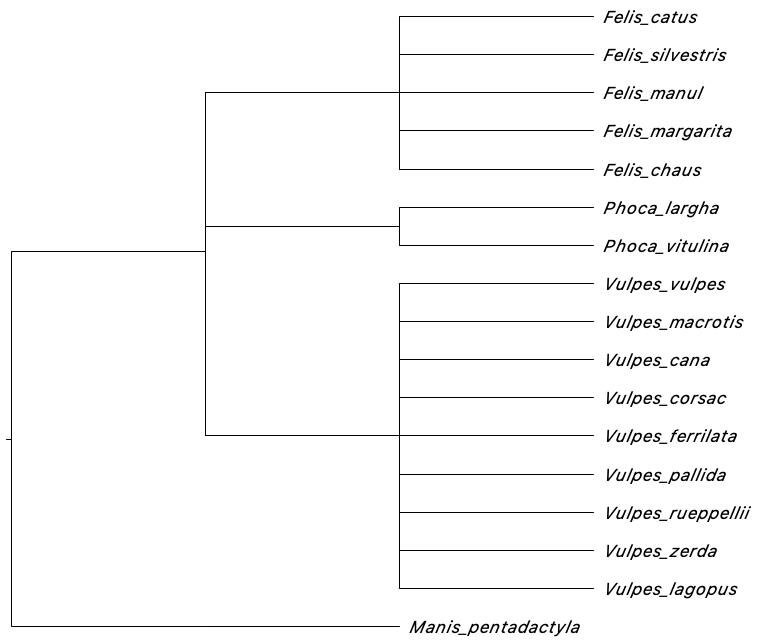
tree.constraint(
taxonomy_folder = "1.CuratedSequences",
targetColumns = c("kingdom", "phylum", "class", "order",
"family", "genus", "species_names"),
Topology = "((ingroup), outgroup);",
outgroup = "Manis_pentadactyla"
)
Particular clades
In this constraint type, phruta will create a constraint
tree for particular clades. For instance, let’s assume that we only need
to create a tree constraining the monophyly within two genera and their
sister relationships:
tree.constraint( taxonomy_folder = "1.CuratedSequences",
targetColumns = c("kingdom", "phylum", "class",
"order", "family", "genus", "species_names"),
Topology = "((Felis), (Phoca));"
)Note that the key aspect in here is the Topology
argument. It is a newick tree.
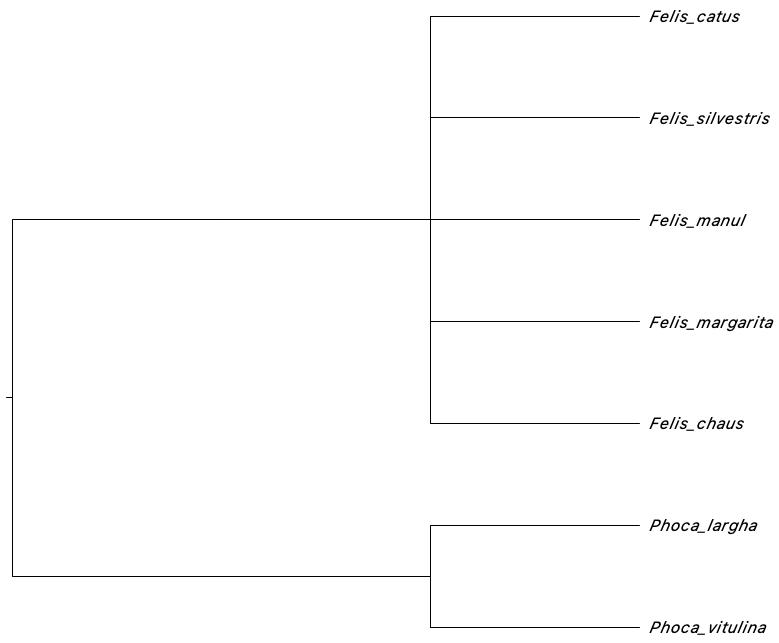
Running PartitionFinder in phruta
With the current version of phruta, users are able to
run PartitionFinder v1 within R. For this,
users should provide the name of the folder where the alignments are
stored, a particular pattern in the file names (masked in
our case), and which models will be run in PartitionFinder.
This function will download PartitionFinder, generate the
input files, and run it all within R. The output files will be in a new
folder within the working directory.
sq.partitionfinderv1(folderAlignments = "2.Alignments",
FilePatterns = "Masked",
models = "all"
)Unfortunately, the output files are not integrated with the current
phruta pipeline. This will be part of a new release.
However, users can still perform gene-based partitioned analyses within
RAxML or can use PartitionFinder’s output files to inform
their own analyses outside phruta.
Partitioned analyses in RAxML
Users can now run partitioned analyses in RAxML within
phruta. This approach is implemented by setting the
partitioned argument in tree.raxml to
TRUE. For now, partitions are based on the genes are being
analyzed. The same model is used to analyze each partition. More details
on partitioned analyses can be customized by passing arguments in
ips::raxml.
tree.raxml(folder = "2.Alignments", FilePatterns = "Masked",
raxml_exec = "raxmlHPC", Bootstrap = 100,
outgroup = "Manis_pentadactyla",
partitioned=T
)Identifying rogue taxa
phruta can help users run RogueNaRok
implemented in the Rogue R package. Users can then examine
whether rogue taxa should be excluded from the analyses.
tree.roguetaxa() uses the bootstrap trees generated using
the tree.raxml() function along with the associated best
tree to identify rogue taxa.
tree.roguetaxa(folder = "3.Phylogeny")