Table of Contents
- Installing RAxML
- Installing PATHd-8 and treePL
- Phylogenetic inference with
phrutaandRAxML - Tree dating in
phruta
Installing RAxML
In MacOS, RAxML can be easily installed to
the PATH using one of the two lines below in
conda:
For other OS (Windows, Linux), please follow the
instructions listed in the official RAxML website
Once RAxML has been installed to your computer, open
R and make sure that the following line doesn’t throw an
error.
system("raxmlHPC")Depending on how RAxML was installed, you may want to
check if RAxML is called from the terminal using
raxmlHPC or raxmlHPC. This string needs to be
passed to tree.raxml() using the argument
raxml_exec. Please note that this argument corresponds to
the exec argument in ips::raxml().
Finally, note that RStudio sometimes has issues finding
stuff in the path while using system(). If you’re
using macOS, try starting RStudio from the
command line by running the following line:
In other OS, it might be better to simply avoid using
RStudio if you’re interested in running the phylogenetic
functions in phruta.
Installing PATHd-8 and treePL
There are excellent guides for installing PATHd-8 and
treePL. Here, I summarize two potentially relevant
options.
First, you can use Brian
O’Meara’s approach for installing PATHd-8 in MacOs and
linux. I summarize the code in the following link.
For Windows users, please use the compiled version of the software
provided in the following link.
Second, you can use homebrew to install treePL (Windows,
MacOS, and Linux), thanks to Jonathan Chang.
Please check the following link) if you’re
interested in running brew from Windows and Linux.
Phylogenetic inference with phruta and
RAxML
Phylogenetic inference is conducted using the
tree.raxml() function. We need to indicate where the
aligned sequences are located (folder argument), the
patterns of the files in the same folder (FilePatterns
argument; “Masked_” in our case). We’ll run a total of 100
boostrap replicates and set the outgroup to “Manis_pentadactyla”.
tree.raxml(folder='2.Alignments',
FilePatterns= 'Masked_',
raxml_exec='raxmlHPC',
Bootstrap=100,
outgroup ="Manis_pentadactyla")The trees are saved in 3.Phylogeny. Likely, the
bipartitions tree, “RAxML_bipartitions.phruta”, is the most relevant.
3.Phylogeny also includes additional
RAxML-related input and output files.
Finally, let’s perform tree dating in our phylogeny using secondary
calibrations extracted from Scholl
and Wiens (2016). This study curated potentially the most
comprenhensive and reliable set of trees to summarize the temporal
dimension in evolution across the tree of life. In phruta,
the trees from Scholl and Wiens (2016) were renamed to match taxonomic
groups.
Tree dating in phruta
Tree dating is performed using the tree.dating()
function in phruta. We have to provide the name of the
folder containing the 1.Taxonomy.csv file created in
sq.curate(). We also have to indicate the name of the
folder containing the RAxML_bipartitions.phruta file. We
will scale our phylogeny using treePL.
tree.dating(taxonomyFolder="1.CuratedSequences",
phylogenyFolder="3.Phylogeny",
scale='treePL')Running this line will result in a new folder
4.Timetree, including the different time-calibrated
phylogenies obained (if any) and associated secondary calibrations used
in the analyses. We found only a few overlapping calibration points
(family-level constraints):
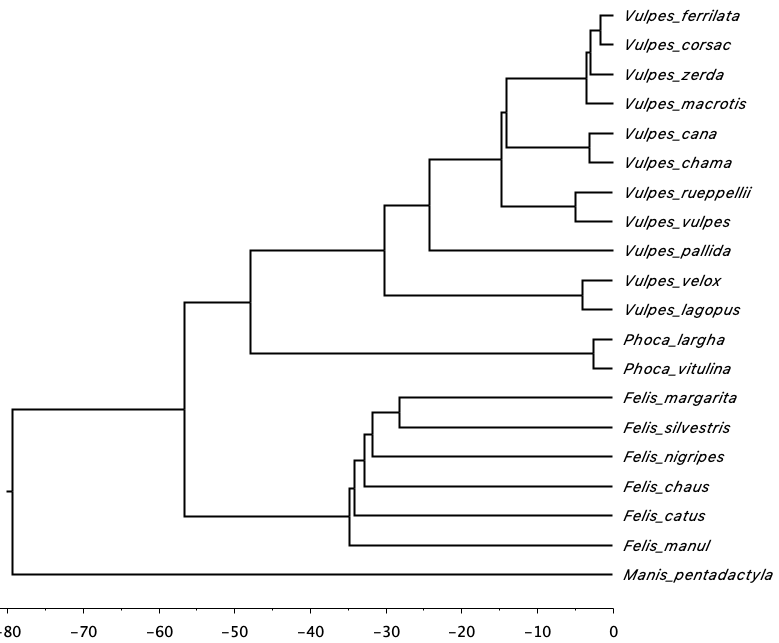
Here’s the resulting time-calibrated phylogeny. The whole process took ~20 minutes to complete on my computer (16 gb RAM, i5).