CANAPE stands for “Categorical Analysis of Neo- And Paleo-Endemism”, and provides insight into the evolutionary processes underlying endemism (Mishler et al. 2014). The idea is basically that endemic regions may be so because either they contain range-restricted parts of a phylogeny that have unusually long branch lengths (paleoendemism), or unusually short branch lengths (neoendemism), or a mixture of both. Paleoendemism may reflect old lineages that have survived extinctions; neoendemism may reflect recently speciated lineages that have not yet dispersed.
This vignette replicates the analysis of Mishler et al. 2014, where CANAPE was originally defined.
Dataset
The canaper package comes with the dataset used in Mishler et al. (2014). Let’s load the data into
memory:
data(acacia)The acacia dataset is a list including two items. The
first, phy, is a phylogeny of Acacia species in
Australia:
acacia$phy
#>
#> Phylogenetic tree with 510 tips and 509 internal nodes.
#>
#> Tip labels:
#> Pararchidendron_pruinosum, Paraserianthes_lophantha, adinophylla, semicircinalis, aphanoclada, inaequilatera, ...
#>
#> Rooted; includes branch lengths.The second, comm, is a community dataframe with species
as columns and rows as sites. The row names (sites) correspond to the
centroids of 50 x 50 km grid cells covering Australia. The community
matrix is too large to print out in its entirety, so we will just take a
look at the first 8 rows and columns1:
dim(acacia$comm)
#> [1] 3037 508
acacia$comm[1:8, 1:8]
#> abbreviata acanthaster acanthoclada acinacea aciphylla acoma acradenia acrionastes
#> -1025000:-1825000 0 0 0 0 0 0 0 0
#> -1025000:-1875000 0 0 0 0 0 0 0 0
#> -1025000:-1925000 0 0 0 0 0 0 0 0
#> -1025000:-1975000 0 0 0 0 0 0 0 0
#> -1025000:-2025000 0 0 0 0 0 0 0 0
#> -1025000:-2075000 0 0 0 0 0 0 0 0
#> -1025000:-2125000 0 0 0 0 0 0 0 0
#> -1025000:-2225000 0 0 0 0 0 0 0 0Randomization test
There are many metrics that describe the phylogenetic diversity of ecological communities. But how do we know if a given metric is statistically significant? One way is with a randomization test. The general process is:
- Generate a set of random communities
- Calculate the metric of interest for each random community
- Compare the observed values to the random values
Observed values that are in the extremes (e.g, the top or lower 5% for a one-sided test, or either the top or bottom 2.5% for a two-sided test) would be considered significantly more or less diverse than random.
The main purpose of canaper is to perform these
randomization tests.
canaper generates random communities using the vegan
package. There are a large number of pre-defined randomization
algorithms available in vegan2, as well as an option
to provide a user-defined algorithm. Selecting the appropriate algorithm
is not trivial, and can greatly influence results3. For details about the
pre-defined algorithms, see vegan::commsim().
This example also demonstrates one of the strengths of
canaper: the ability to run randomizations in parallel4. This is
by far the most time-consuming part of CANAPE, since we have to repeat
the calculations many (e.g., hundreds or more) times across the
randomized communities to obtain reliable results. Here, we set the
number of iterations (n_iterations; i.e., the number of
swaps used to produce each randomized community) fairly high because
this community matrix is large and includes many zeros; thorough mixing
by swapping many times is required to completely randomize the
matrix.
We will use a low number of random communities (n_reps)
so things finish relatively quickly; you should consider increasing
n_reps for a “real” analysis5. We will use the
curveball randomization algorithm, which maintains species
richness and abundance patterns while randomizing species identity (Strona et al. 2014)6.
# Set a parallel back-end, with 2 CPUs running simultaneously
plan(multisession, workers = 2)
# Uncomment this to show a progress bar when running cpr_rand_test()
# progressr::handlers(global = TRUE) # nolint
# Set a random number generator seed so we get the same results if this is
# run again
set.seed(071421)
tic() # Set a timer
# Run randomization test
acacia_rand_res <- cpr_rand_test(
acacia$comm, acacia$phy,
null_model = "curveball",
n_reps = 20, n_iterations = 100000,
tbl_out = TRUE
)
#> Warning: Abundance data detected. Results will be the same as if using presence/absence data (no abundance weighting is used).
#> Warning: Dropping tips from the tree because they are not present in the community data:
#> Pararchidendron_pruinosum, Paraserianthes_lophantha
toc() # See how long it took
#> 70.204 sec elapsed
# Switch back to sequential (non-parallel) mode
plan(sequential)Let’s take a peek at the output.
acacia_rand_res
#> # A tibble: 3,037 × 55
#> site pd_obs pd_ra…¹ pd_ra…² pd_ob…³ pd_ob…⁴ pd_ob…⁵ pd_ob…⁶ pd_ob…⁷ pd_ob…⁸ pd_al…⁹ pd_al…˟ pd_al…˟ pd_al…˟ pd_al…˟ pd_al…˟ pd_al…˟ pd_al…˟ pd_al…˟ rpd_obs rpd_r…˟ rpd_r…˟ rpd_o…˟ rpd_o…˟ rpd_o…˟ rpd_o…˟ rpd_o…˟ rpd_o…˟ pe_obs pe_ra…˟ pe_ra…˟ pe_ob…˟ pe_ob…˟ pe_ob…˟ pe_ob…˟ pe_ob…˟ pe_ob…˟
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 -1025000… 0.0145 0.0207 0.00432 -1.46 1 19 20 0.05 0.95 0.0355 0.0414 0.00779 -0.753 5 14 20 0.25 0.7 0.407 0.514 0.129 -0.827 4 16 20 0.2 0.8 2.47e-5 6.14e-5 5.92e-5 -0.619 3 17 20 0.15 0.85
#> 2 -1025000… 0.0382 0.0522 0.00704 -1.99 0 20 20 0 1 0.0680 0.0883 0.00651 -3.10 0 20 20 0 1 0.561 0.595 0.0944 -0.358 9 11 20 0.45 0.55 1.44e-4 2.82e-4 1.86e-4 -0.738 5 15 20 0.25 0.75
#> 3 -1025000… 0.0378 0.0371 0.00535 0.130 12 8 20 0.6 0.4 0.0572 0.0631 0.00797 -0.743 2 17 20 0.1 0.85 0.661 0.593 0.0941 0.724 16 4 20 0.8 0.2 1.71e-4 2.42e-4 1.62e-4 -0.439 7 13 20 0.35 0.65
#> 4 -1025000… 0.0570 0.0606 0.00758 -0.480 6 14 20 0.3 0.7 0.0858 0.104 0.0105 -1.77 0 20 20 0 1 0.664 0.586 0.0913 0.863 17 3 20 0.85 0.15 5.30e-4 3.56e-4 2.33e-4 0.748 17 3 20 0.85 0.15
#> 5 -1025000… 0.0409 0.0423 0.00836 -0.176 11 9 20 0.55 0.45 0.0542 0.0713 0.00904 -1.89 0 19 20 0 0.95 0.753 0.596 0.102 1.55 19 1 20 0.95 0.05 1.25e-4 2.20e-4 1.31e-4 -0.728 6 14 20 0.3 0.7
#> 6 -1025000… 0.00998 0.0102 0.00206 -0.0890 11 9 20 0.55 0.45 0.0276 0.0173 0.00742 1.39 18 2 20 0.9 0.1 0.361 0.695 0.337 -0.989 2 18 20 0.1 0.9 1.17e-5 3.86e-5 4.82e-5 -0.557 3 17 20 0.15 0.85
#> 7 -1025000… 0.0187 0.0230 0.00378 -1.14 3 17 20 0.15 0.85 0.0325 0.0390 0.00820 -0.782 2 16 20 0.1 0.8 0.575 0.619 0.189 -0.231 11 9 20 0.55 0.45 5.20e-5 9.07e-5 6.56e-5 -0.589 5 15 20 0.25 0.75
#> 8 -1025000… 0.0434 0.0516 0.00862 -0.955 4 16 20 0.2 0.8 0.0779 0.0956 0.00995 -1.78 1 19 20 0.05 0.95 0.557 0.544 0.100 0.125 12 8 20 0.6 0.4 4.32e-4 3.14e-4 1.24e-4 0.944 16 4 20 0.8 0.2
#> 9 -1025000… 0.0111 0.0104 0.00172 0.418 17 3 20 0.85 0.15 0.0168 0.0211 0.00715 -0.607 6 14 20 0.3 0.7 0.662 0.558 0.227 0.461 16 4 20 0.8 0.2 2.73e-5 1.94e-5 1.53e-5 0.521 17 3 20 0.85 0.15
#> 10 -1025000… 0.0903 0.0896 0.0114 0.0601 13 7 20 0.65 0.35 0.115 0.143 0.00775 -3.51 0 20 20 0 1 0.783 0.629 0.0760 2.02 19 1 20 0.95 0.05 5.07e-4 5.76e-4 2.10e-4 -0.328 10 10 20 0.5 0.5
#> # … with 3,027 more rows, 18 more variables: pe_alt_obs <dbl>, pe_alt_rand_mean <dbl>, pe_alt_rand_sd <dbl>, pe_alt_obs_z <dbl>, pe_alt_obs_c_upper <dbl>, pe_alt_obs_c_lower <dbl>, pe_alt_obs_q <dbl>, pe_alt_obs_p_upper <dbl>, pe_alt_obs_p_lower <dbl>, rpe_obs <dbl>, rpe_rand_mean <dbl>,
#> # rpe_rand_sd <dbl>, rpe_obs_z <dbl>, rpe_obs_c_upper <dbl>, rpe_obs_c_lower <dbl>, rpe_obs_q <dbl>, rpe_obs_p_upper <dbl>, rpe_obs_p_lower <dbl>, and abbreviated variable names ¹pd_rand_mean, ²pd_rand_sd, ³pd_obs_z, ⁴pd_obs_c_upper, ⁵pd_obs_c_lower, ⁶pd_obs_q, ⁷pd_obs_p_upper, ⁸pd_obs_p_lower,
#> # ⁹pd_alt_obs, ˟pd_alt_rand_mean, ˟pd_alt_rand_sd, ˟pd_alt_obs_z, ˟pd_alt_obs_c_upper, ˟pd_alt_obs_c_lower, ˟pd_alt_obs_q, ˟pd_alt_obs_p_upper, ˟pd_alt_obs_p_lower, ˟rpd_rand_mean, ˟rpd_rand_sd, ˟rpd_obs_z, ˟rpd_obs_c_upper, ˟rpd_obs_c_lower, ˟rpd_obs_q, ˟rpd_obs_p_upper, ˟rpd_obs_p_lower,
#> # ˟pe_rand_mean, ˟pe_rand_sd, ˟pe_obs_z, ˟pe_obs_c_upper, ˟pe_obs_c_lower, ˟pe_obs_q, ˟pe_obs_p_upper, ˟pe_obs_p_lowercpr_rand_test() produces a lot of
columns. Here, pd_obs is the observed value of phylogenetic
diversity (PD). Other columns starting with pd refer to
aspects of the randomization: pd_rand_mean is the mean PD
across the random communities, pd_rand_sd is the standard
deviation of PD across the random communities, pd_obs_z is
the standard effect size of PD, etc.
For details about what each column means, see
cpr_rand_test().
Classify endemism
The next step in CANAPE is to classify types of endemism. For a full
description, see Mishler et al. (2014). In
short, this defines endemic regions based on combinations of the
p-values of phylogenetic endemism (PE; pe_obs) and
PE measured on an alternative tree with all branch lengths equal
(pe_alt). Here is a summary borrowed
from the biodiverse blog, modified to use the variables as they are
defined in canaper:
- If either
pe_obsorpe_alt_obsare significantly high then we look for paleo- or neo-endemism- If
rpe_obsis significantly high then we have palaeo-endemism - Else if
rpe_obsis significantly low then we have neo-endemism - Else we have mixed age endemism, in which case
- If both
pe_obsandpe_alt_obsare highly significant (p < 0.01) then we have super endemism (high in both paleo and neo) - Else we have mixed (some mixture of paleo, neo and nonendemic)
- If both
- If
- Else if neither
pe_obsorpe_alt_obsare significantly high then we have a non-endemic cell
cpr_classify_endem() carries this out automatically on
the results from cpr_rand_test(), adding a factor called
endem_type:
acacia_canape <- cpr_classify_endem(acacia_rand_res)
table(acacia_canape$endem_type)
#>
#> mixed neo not significant paleo super
#> 99 12 2783 68 75Classify significance
A similar function to cpr_classify_endem() is available
to classify significance of the randomization test,
cpr_classify_signif(). Note that both of these take a
data.frame as input and return a data.frame as
output, so they are “pipe-friendly”. The second argument of
cpr_classify_signif() is the name of the biodiversity
metric that you want to classify. This will add a column
*_signif with the significance relative to the random
distribution for that metric. For example,
cpr_classify_signif(df, "pd") will add the
pd_signif column to df.
We can chain them together as follows:
acacia_canape <-
cpr_classify_endem(acacia_rand_res) |>
cpr_classify_signif("pd") |>
cpr_classify_signif("rpd") |>
cpr_classify_signif("pe") |>
cpr_classify_signif("rpe")
# Take a look at one of the significance classifications:
table(acacia_canape$pd_signif)
#>
#> < 0.01 > 0.99 not significant
#> 582 91 2364Visualize results
With the randomizations and classification steps taken care of, we can now visualize the results to see how they match up with those of Mishler et al. (2014).
Note that the results will not be identical because we have used a different randomization algorithm from the paper and because of stochasticity in the random values, as well as fewer replicates for the randomization test.
Here is Figure 2, showing the results of the randomization test for PE, RPE, PE, and RPE:
# Fist do some data wrangling to make the results easier to plot
# (add lat/long columns)
acacia_canape <- acacia_canape |>
separate(site, c("long", "lat"), sep = ":") |>
mutate(across(c(long, lat), parse_number))
theme_update(
panel.background = element_rect(fill = "white", color = "white"),
panel.grid.major = element_line(color = "grey60"),
panel.grid.minor = element_blank()
)
a <- ggplot(acacia_canape, aes(x = long, y = lat, fill = pd_signif)) +
geom_tile() +
# cpr_signif_cols_2 is a CVD-friendly color palette in canaper
scale_fill_manual(values = cpr_signif_cols_2, name = "Phylogenetic diversity") +
guides(fill = guide_legend(title.position = "top", label.position = "bottom"))
b <- ggplot(acacia_canape, aes(x = long, y = lat, fill = rpd_signif)) +
geom_tile() +
scale_fill_manual(
values = cpr_signif_cols_2, name = "Relative phylogenetic diversity"
) +
guides(fill = guide_legend(title.position = "top", label.position = "bottom"))
c <- ggplot(acacia_canape, aes(x = long, y = lat, fill = pe_signif)) +
geom_tile() +
scale_fill_manual(values = cpr_signif_cols_2, name = "Phylogenetic endemism") +
guides(fill = guide_legend(title.position = "top", label.position = "bottom"))
d <- ggplot(acacia_canape, aes(x = long, y = lat, fill = rpe_signif)) +
geom_tile() +
scale_fill_manual(
values = cpr_signif_cols_2, name = "Relative phylogenetic endemism"
) +
guides(fill = guide_legend(title.position = "top", label.position = "bottom"))
a + b + c + d +
plot_annotation(tag_levels = "a") & theme(legend.position = "top")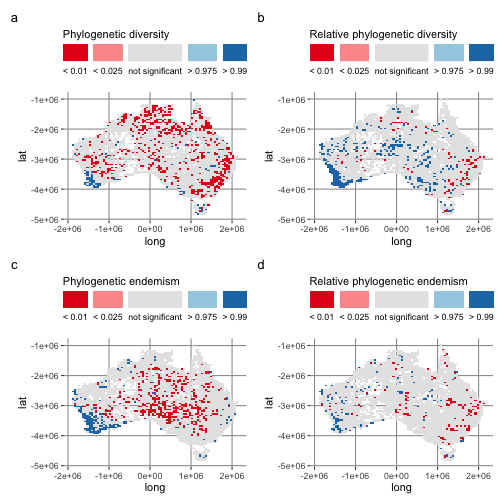
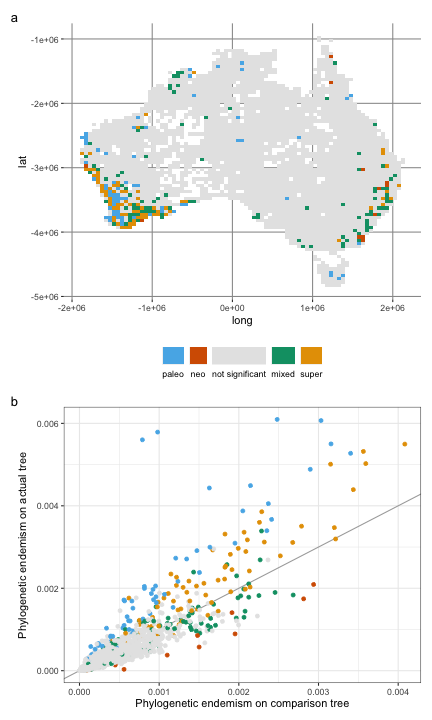
And here is Figure 3, showing the results of CANAPE:
a <- ggplot(acacia_canape, aes(x = long, y = lat, fill = endem_type)) +
geom_tile() +
# cpr_endem_cols_4 is a CVD-friendly color palette in canaper
scale_fill_manual(values = cpr_endem_cols_4) +
guides(
fill = guide_legend(title.position = "top", label.position = "bottom")
) +
theme(legend.position = "bottom", legend.title = element_blank())
b <- ggplot(
acacia_canape,
aes(x = pe_alt_obs, y = pe_obs, color = endem_type)
) +
geom_abline(slope = 1, color = "darkgrey") +
geom_point() +
scale_color_manual(values = cpr_endem_cols_4) +
labs(
x = "Phylogenetic endemism on comparison tree",
y = "Phylogenetic endemism on actual tree"
) +
theme_bw() +
theme(legend.position = "none")
a + b + plot_layout(ncol = 1) + plot_annotation(tag_levels = "a")