How many randomizations?
Joel H. Nitta
05 April, 2023
Source:vignettes/how-many-rand.Rmd
how-many-rand.RmdA central part of CANAPE is comparing observed values with randomizations. It is up to the user to set the number of randomizations… but how does one know if the number of randomizations is sufficient?
(This vignette assumes a basic understanding of CANAPE, community data matrices, and randomizations. If you aren’t familiar with any of these, you should probably see the the CANAPE example vignette first).
A word about randomization algorithms
There is a rich literature on choice of randomization algorithms in ecology (see references below), so I won’t go into this too much, but choice of algorithm can have a large impact on results, so it’s important to understand.
As described by Strona et al. (2018), randomization algorithms can vary in their degree of degree of conservation; that is, how closely they resemble the original data. At one extreme, some algorithms require row and column sums (i.e., marginal sums) to be perfectly preserved between the original data and the randomized data (more conservative)1; at the other, marginal sums may be completely different (less conservative). More conservative algorithms are less prone to type II error but more prone to type I error; the opposite is true for less conservative algorithms. Generally you want the randomization algorithm to only change the one aspect of the data that you are interested in testing. The ultimate choice of algorithm will depend on the data, ecological conditions, and computing restraints.
canaper uses the vegan
package for randomizations. There are a large number of pre-defined
randomization algorithms available in vegan2, as well as the option
to provide a user-defined algorithm. For details about each pre-defined
algorithm, see vegan::commsim().
For this vignette, I will use the swap algorithm, a
“more conservative” algorithm that preserves marginal sums (Gotelli and Entsminger 2003) and has been
widely used in ecological studies3.
Replicates vs. iterations
Before proceeding, we need to clarify some terminology.
cpr_rand_test() includes two arguments, n_reps
and n_iterations. These sound similar but refer to two very
different things.
n_reps is the number of random communities to simulate.
For example, if n_reps is 100, will we be comparing each
observed value (e.g., phylogenetic diversity, pd_obs), with
100 random replicates of pd_obs. If n_reps is
too low, we will lack sufficiently statistical power to detect patterns
in the data.
n_iterations is only used by some randomization
algorithms, the “sequential” algorithms. Sequential algorithms randomize
a community matrix by exchanging values between existing cells
(“swapping”). As you might guess, the swap algorithm is a
sequential algorithm. One such swap is a single “iteration”. If the
total number of iterations, n_iterations, is too low, the
randomized matrix won’t be sufficiently randomized, and will still
resemble the original matrix4.
If either n_reps or n_iterations are set
too high, it will take overly long to finish the calculations. So our
goal is to set them sufficiently high to achieve proper randomization,
but not so high cpr_rand_test() never finishes.
Effect of n_iterations
First, let’s load the packages used in this vignette.
library(canaper) # This package
library(tictoc) # For timing
# For data-wrangling and plotting
library(ggplot2)
library(dplyr)Next we will test the effects of n_iterations, using the
test
dataset that comes with canaper (and Biodiverse).
I will compare the percentage similarity between the original matrix and
successive iterations of a randomized matrix. This can be done using the
cpr_iter_sim() function:
# Conduct up to 10,000 iterations (swaps),
# recording similarity every 10 iterations
iter_sim_res <- cpr_iter_sim(
comm = biod_example$comm,
null_model = "swap",
n_iterations = 10000,
thin = 10,
seed = 123
)
# Check the output
iter_sim_res
#> # A tibble: 1,000 × 2
#> iteration similarity
#> <int> <dbl>
#> 1 10 0.990
#> 2 20 0.981
#> 3 30 0.973
#> 4 40 0.965
#> 5 50 0.957
#> 6 60 0.949
#> 7 70 0.944
#> 8 80 0.938
#> 9 90 0.934
#> 10 100 0.928
#> # … with 990 more rows
# Plot the results
ggplot(iter_sim_res, aes(x = iteration, y = similarity)) +
geom_line() +
labs(x = "Num. iterations", y = "% Similarity")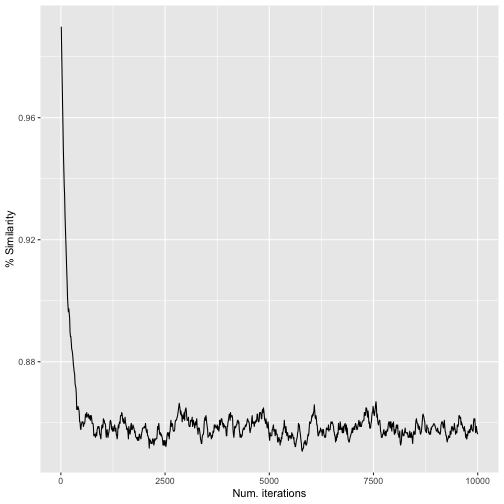
iter_sim_res() resultsFrom this, we can see that the original community and the randomized community reach a maximum dissimilarity at ca. 500–1,000 iterations. After that, the randomized community doesn’t become any more different with additional “mixing”.
Note that the number of iterations required will vary based on the dataset. Large matrices with many zeros will likely take more iterations, and even then still retain relatively high similarity between the original matrix and the randomized matrix. So I recommend exploring the data as above to determine the minimum number of iterations needed.
Now that we’ve settled on the number of iterations per random replicate (1,000), let’s look into the number of replicates.
Effect of n_reps
With randomizations, there is no “right” answer, so we can’t test to
see that cpr_rand_test() produces the exact answer we’re
looking for. Rather, we will check that it starts to converge on
approximately the same result once n_reps is high
enough.
Here, I will compare the percentile of observed phylogenetic
diversity relative to random (pd_obs_p_upper, one
of the values used for calculating endemism type) between pairs of
random communities each generated with the same number of replicates5. I will
also time calculations for one of each pair.
# Specify a different random seed for each set of randomizations so they give
# different, reproducible results
# First set (time these)
set.seed(12345)
tic()
res_10_1 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 10, tbl_out = TRUE
)
toc()
#> 2.681 sec elapsed
tic()
res_100_1 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 100, tbl_out = TRUE
)
toc()
#> 3.672 sec elapsed
tic()
res_1000_1 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 1000, tbl_out = TRUE
)
toc()
#> 15.465 sec elapsed
# Second set
set.seed(67890)
res_10_2 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 10, tbl_out = TRUE
)
res_100_2 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 100, tbl_out = TRUE
)
res_1000_2 <- cpr_rand_test(
biod_example$comm, biod_example$phy,
null_model = "swap",
n_iterations = 1000, n_reps = 1000, tbl_out = TRUE
)Next, plot the results.
# We will make the same plot repeatedly, so write
# a quick function to avoid lots of copying and pasting
plot_comp <- function(res_1, res_2) {
left_join(
select(res_1, site, pd_obs_p_upper_1 = pd_obs_p_upper),
select(res_2, site, pd_obs_p_upper_2 = pd_obs_p_upper),
by = "site"
) |>
ggplot(aes(x = pd_obs_p_upper_1, y = pd_obs_p_upper_2)) +
geom_point() +
geom_abline(slope = 1, intercept = 0)
}
plot_comp(res_10_1, res_10_2) + labs(title = "10 replicates")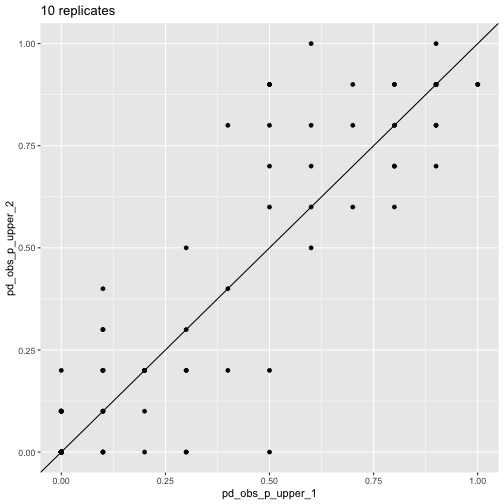
pd_obs_p_upper across
runs, 10 reps
plot_comp(res_100_1, res_100_2) + labs(title = "100 replicates")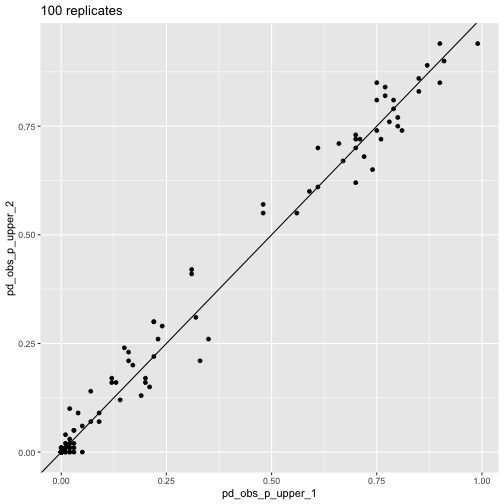
pd_obs_p_upper across
runs, 100 reps
plot_comp(res_1000_1, res_1000_2) + labs(title = "1000 replicates")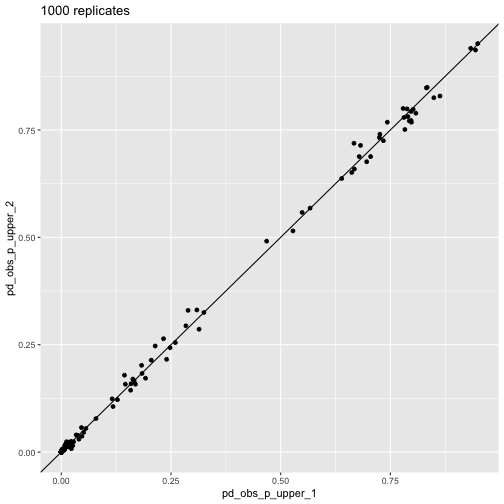
pd_obs_p_upper across
runs, 1,000 repsThis visualization shows how the randomization results converge as
n_reps increases.
The above plots illustrate convergence for one particular aspect of CANAPE, but what about endemism type itself? We can look at that too.
# Define a helper function that joins two datasets and calculates % agreement
# on endemism type between them
calc_agree_endem <- function(df_1, df_2, n_reps) {
left_join(
df_1 |> cpr_classify_endem() |> select(site, endem_type_1 = endem_type),
df_2 |> cpr_classify_endem() |> select(site, endem_type_2 = endem_type),
by = "site"
) |>
mutate(agree = endem_type_1 == endem_type_2) |>
summarize(agree = sum(agree), total = n()) |>
mutate(
p_agree = agree / total,
n_reps = n_reps
)
}
bind_rows(
calc_agree_endem(res_10_1, res_10_2, 10),
calc_agree_endem(res_100_1, res_100_2, 100),
calc_agree_endem(res_1000_1, res_1000_2, 1000),
)
#> # A tibble: 3 × 4
#> agree total p_agree n_reps
#> <int> <int> <dbl> <dbl>
#> 1 100 127 0.787 10
#> 2 118 127 0.929 100
#> 3 125 127 0.984 1000At 1,000 replicates, we see very high agreement on endemism type between the two randomizations.
Of course, another important consideration is how
long calculations take. You can see that time increases with
n_reps, but not exactly in a linear fashion. We don’t have
the space to go into benchmarking here, but this illustrates the time /
n_reps trade-off6.
Conclusion
In this case (the example
dataset that comes with canaper), we see that a minimum
of 1,000 random replicates with 1,000 swapping iterations per replicate
is probably needed to attain robust results.
I hope this vignette helps you determine the settings to use for your own dataset!