![]()
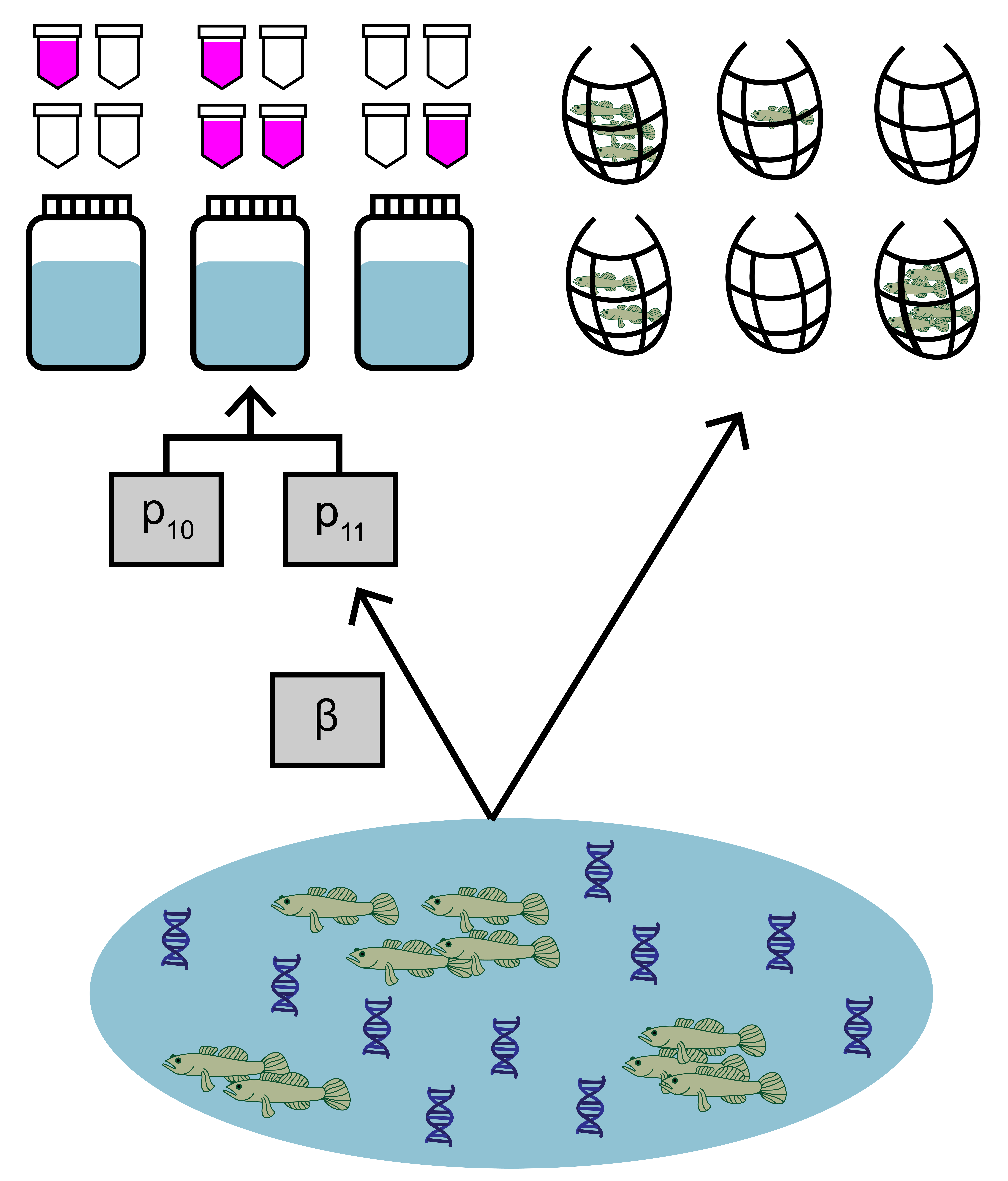
The package eDNAjoint is useful for interpreting observations from paired or semi-paired environmental DNA (eDNA) and traditional surveys. The package runs a Bayesian model that integrates these two data streams to jointly estimate parameters like the false positive probability of eDNA detection and expected catch rate at a site. Optional model variations allow inclusion of site-level covariates that scale the sensitivity of eDNA sampling relative to traditional sampling, as well as estimation of gear scaling coefficients when multiple traditional gear types are used. Additional functions in the package facilitate interpretation of model fits.
Check out a paper about the package in Methods in Ecology and Evolution.
Installation
The most stable version of eDNAjoint can be found on Cran:
install.packages("eDNAjoint")You can also install the development version of eDNAjoint from ROpenSci:
install.packages("eDNAjoint", repos = "https://ropensci.r-universe.dev")Example
The main functionality in eDNAjoint is the use of joint_model() that will fit the model to data. Further functions like joint_summarize() and detection_calculate() can be used to help with model fit interpretation.
This example fits the joint model to data from paired, replicated eDNA qPCR and seine sampling observations of endangered tidewater gobies (Eucyclogobius newberryi) from a study by Schmelzle and Kinziger (2016). The following variation of the joint model includes site-level covariates that scale the sensitivity of eDNA sampling relative to traditional sampling.
library(eDNAjoint)
data(goby_data)
# run the joint model with two covariates
goby_fit <- joint_model(data = goby_data, cov = c("Filter_time", "Salinity"),
family = "poisson", p10_priors = c(1, 20), q = FALSE)And then this model fit can be accessed to do things like summarize the posterior distribution for the probability of a false positive detection, :
# summarize p10 posterior
joint_summarize(goby_fit$model, par = "p10")
#> mean se_mean sd 2.5% 97.5% n_eff Rhat
#> p10 0.003 0 0.001 0.001 0.007 13482.83 1Or to find the number of eDNA samples and traditional survey samples necessary to detect presence of the species at a given expected catch rate:
# find the number of samples necessary to detect presence with 0.9 probability
# at the mean covariate values, if the expected catch rate (mu) is 0.1, 0.5, or
# 1 individuals/traditional survey unit.
detection_calculate(goby_fit$model, mu = c(0.1, 0.5, 1),
cov_val = c(0, 0), probability = 0.9)
#> mu n_traditional n_eDNA
#> [1,] 0.1 24 14
#> [2,] 0.5 5 4
#> [3,] 1.0 3 2Vignette
You can find much more detailed examples of the functions in eDNAjoint and the model underlying the package in the package vignette.
Citation
Using eDNAjoint in a manuscript? Please consider citing either the manuscript about the package:
Keller, A.G., & Kelly, R.P. (2025). eDNAjoint: An R package for interpreting paired or semi-paired environmental DNA and traditional survey data in a Bayesian framework. Methods in Ecology and Evolution, 00, 1–9. https://doi.org/10.1111/2041-210X.70000
Contributing
Interested in contributing to this package? See some notes on contributing.
Code of Conduct
Please note that eDNAjoint is released with a Contributor Code of Conduct. By contributing to this project you agree to abide by its terms.
References
Keller, A.G., & Kelly, R.P. (2025). eDNAjoint: An R package for interpreting paired or semi-paired environmental DNA and traditional survey data in a Bayesian framework. Methods in Ecology and Evolution, 00, 1–9. https://doi.org/10.1111/2041-210X.70000
Keller, A.G., Grason, E.W., McDonald, P.S., Ramon-Laca, A., Kelly, R.P. (2022). Tracking an invasion front with environmental DNA. Ecological Applications. 32(4): e2561. https://doi.org/10.1002/eap.2561
Schmelzle, M.C. and Kinziger, A.P. (2016). Using occupancy modelling to compare environmental DNA to traditional field methods for regional-scale monitoring of an endangered aquatic species. Molecular Ecology Resources. 16(4): 895-908. https://doi.org/10.1111/1755-0998.12501