Extending NeXML: an example based on simmap
Carl Boettiger
Scott Chamberlain
Rutger Vos
Hilmar Lapp
Source:vignettes/simmap.Rmd
simmap.RmdExtending the NeXML standard through metadata annotation.
Here we illustrate this process using the example of stochastic character mapping (Huelsenbeck et al. 2003). A stochastic character map is simply an annotation of the branches on a phylogeny, assigning each section of each branch to a particular “state” (typically of a morphological characteristic).
Bollback (2006) provides a widely used
stand-alone software implementation of this method in the software
simmap, which modified the standard Newick tree format to
express this additional information. This can break compatibility with
other software, and creates a format that cannot be interpreted without
additional information describing this convention. By contrast, the
NeXML extension is not only backwards compatible but contains a precise
and machine-readable description of what it is encoding.
In this example, we illustrate how the additional information
required to define a stochastic character mapping (a simmap
mapping) in NeXML.
Revell (2012) describes the
phytools package for R, which includes utilities for
reading, manipulating, and writing simmap files in R. In
this example, we also show how to define RNeXML functions
that map the R representation used by Revell (an extension of the
ape class) into the NeXML extension we have defined by
using RNeXML functions.
Since a stochastic character map simply assigns different states to
parts of a branch (or edge) on the phylogenetic tree, we can create a
NeXML representation by annotating the edge elements with
appropriate meta elements. These elements need to describe
the character state being assigned and the duration (in terms of
branch-length) that the edge spends in that state (Stochastic character
maps are specific to time-calibrated or ultrametric trees).
NeXML already defines the characters element to handle
discrete character traits (nex:char) and the states they
can assume (nex:state). We will thus reuse the
characters element for this purpose, referring to both the
character trait and the states by the ids assigned to them in that
element. (NeXML’s convention of referring to everything by id permits a
single canonical definition of each term, making it clear where
additional annotation belongs). For each edge, we need to indicate:
- That our annotation contains a stochastic character mapping reconstruction
- Since many reconstructions are possible for a single edge, we give each reconstruction an id
- We indicate for which character trait we are defining the reconstruction
- We then indicate which states the character assumes on that edge.
For each state realized on the edge, that involves stating:
- the state assignment
- the duration (length of time) for which the edge spends in the given state
- the order in which the state changes happen (Though we could just assume state transitions are listed chronologically, NeXML suggests making all data explicit, rather than relying on the structure of the data file to convey information).
Thus the annotation for an edge that switches from state
s2 to state s1 of character cr1
would be constructed like this:
m <- meta("simmap:reconstructions", children = c(
meta("simmap:reconstruction", children = c(
meta("simmap:char", "cr1"),
meta("simmap:stateChange", children = c(
meta("simmap:order", 1),
meta("simmap:length", "0.2030"),
meta("simmap:state", "s2"))),
meta("simmap:char", "cr1"),
meta("simmap:stateChange", children = c(
meta("simmap:order", 2),
meta("simmap:length", "0.0022"),
meta("simmap:state", "s1")))
))))Of course writing out such a definition manually becomes tedious
quickly. Because these are just R commands, we can easily define a
function that can loop over an assignment like this for each edge,
extracting the appropriate order, length and state from an existing R
object such as that provided in the phytools package.
Likewise, it is straightforward to define a function that reads this
data using the RNeXML utilities and converts it back to the
phytools package. The full implementation of this mapping
can be seen in the simmap_to_nexml() and the
nexml_to_simmap() functions provided in the
RNeXML package.
As the code indicates, the key step is simply to define the data in
meta elements. In so doing, we have defined a custom namespace,
simmap, to hold our variables. This allows us to provide a
URL with more detailed descriptions of what each of these elements
mean:
nex <- add_namespaces(c(simmap = "https://github.com/ropensci/RNeXML/tree/master/inst/simmap.md"))At that URL we have posted a simple description of each term.
Using this convention we can generate NeXML files containing
simmap data, read those files into R, and convert them back
into the phytools package format. These simple functions
serve as further illustration of how RNeXML can be used to
extend the NeXML standard. We illustrate their use briefly here,
starting with loading a nexml object containing a
simmap reconstruction into R:
f <- system.file("examples", "simmap_ex.xml", package = "RNeXML")
simmap_ex <- read.nexml(f)The get_trees() function can be used to return an
ape::phylo tree as usual. RNeXML automatically
detects the simmap reconstruction data and returns includes
this in a maps element of the ape::phylo
object, for use with other phytools functions.
phy <- nexml_to_simmap(simmap_ex)We can then use various functions from phytools designed
for simmap objects (Revell
2012), such as the plotting function:
library("phytools")
plotSimmap(phy)no colors provided. using the following legend:
A B C
"black" "red" "green3" 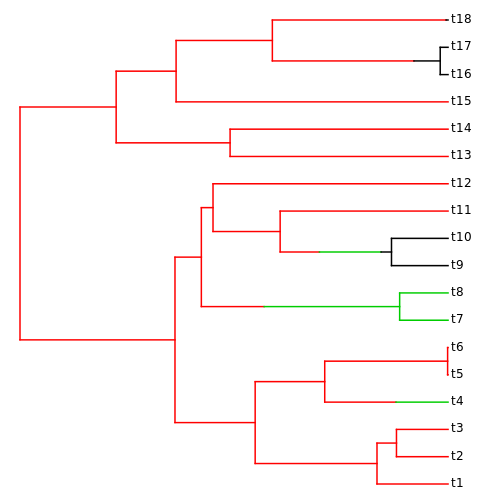
Likewise, we can convert the object back in the NeXML format and write it out to file to be read by other users.
nex <- simmap_to_nexml(phy)
nexml_write(nex, "simmap.xml")[1] "simmap.xml"Though other NeXML parsers (for instance, for Perl or Python) have
not been written explicitly to express simmap data, those
parsers will nonetheless be able to successfully parse this file and
expose the simmap data to the user.